

Cellular senescence is a state of stable and essentially irreversible cell cycle arrest that occurs in response to various forms of cellular stress. Although senescent cells remain metabolically active, they lose the ability to proliferate, often adopting a distinct phenotype that includes changes in gene expression, chromatin structure, and secretory activity.
Unlike quiescent or terminally differentiated cells, senescent cells are defined by their unique molecular and functional hallmarks, including the activation of tumor suppressor pathways such as p53/p21CIP1 and p16INK4a/Rb, and the production of a senescence-associated secretory phenotype (SASP).
The concept of cellular senescence was first introduced by Leonard Hayflick and Paul Moorhead in the early 1960s, when they discovered that human fibroblasts have a limited capacity to divide in culture, a phenomenon now known as the Hayflick limit.
This breakthrough challenged the belief that cultured cells were immortal and introduced the idea that cellular aging was an intrinsic, regulated process. Over the following decades, researchers expanded this concept to include various forms of stress-induced senescence, such as oncogene-induced senescence (OIS) and therapy-induced senescence (TIS), deepening our understanding of senescence as a key component of tumor suppression, tissue remodeling, and age-related pathologies.
In the context of cancer biology, cellular senescence plays a complex and paradoxical role. On one hand, it serves as a critical barrier to malignant transformation by halting the proliferation of damaged or premalignant cells. On the other hand, the accumulation of senescent cells and their pro-inflammatory secretome can fuel a pro-tumorigenic microenvironment, contribute to immune evasion, and even promote metastasis or therapy resistance.
In this blog post, we will explore the molecular mechanisms that govern cellular senescence, examine the key pathways and biomarkers involved, and analyze its dual role in cancer progression and therapy response. We will also discuss the latest research on senescence-targeted therapies and the tools used to study senescence in cancer models.
II. Molecular Mechanisms of Cellular Senescence
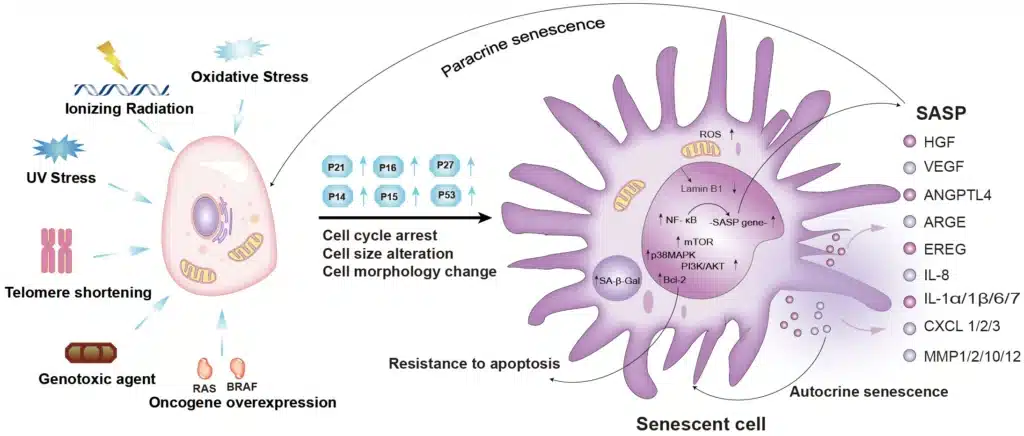
Cellular senescence is governed by a network of complex molecular mechanisms that detect stress signals and enforce a stable cell cycle arrest. The induction and maintenance of this state involve a variety of upstream triggers, signaling cascades, and structural changes that coordinate the cell’s response to damage or oncogenic stimuli.
A. Triggers of Senescence
- Replicative Senescence
This is the earliest discovered form of senescence, caused by telomere attrition. With each cell division, telomeres shorten due to the end-replication problem. When telomeres become critically short, they are recognized as sites of DNA damage, activating the DNA damage response (DDR) and leading to permanent cell cycle arrest. - Oncogene-Induced Senescence (OIS)
Activation of certain oncogenes such as RAS, BRAF, or MYC can paradoxically trigger a strong anti-proliferative response. OIS is characterized by excessive mitogenic signaling, replication stress, and accumulation of DNA damage, which activate tumor suppressor pathways and drive senescence. - DNA Damage-Induced Senescence
Exposure to genotoxic agents such as ionizing radiation, UV light, or chemotherapeutic drugs causes double-strand breaks (DSBs). These lesions activate DDR kinases such as ATM and ATR, initiating senescence to prevent propagation of damaged DNA. - Oxidative Stress and Mitochondrial Dysfunction
Excessive production of reactive oxygen species (ROS) damages cellular macromolecules and organelles, contributing to senescence. Mitochondrial dysfunction-associated senescence (MiDAS) is an emerging subtype characterized by ROS accumulation, metabolic reprogramming, and SASP activation.
B. Key Molecular Pathways
- p53/p21CIP1 Pathway
DNA damage stabilizes p53, a tumor suppressor and transcription factor, which induces p21CIP1, a cyclin-dependent kinase inhibitor. p21 blocks CDK2, maintaining hypophosphorylated Rb and enforcing G1/S cell cycle arrest. This pathway is critical in early senescence induction. - p16INK4a/Rb Pathway
p16INK4a inhibits CDK4/6, leading to sustained hypophosphorylation of retinoblastoma protein (Rb). Hypophosphorylated Rb binds to E2F transcription factors, repressing genes required for S-phase entry and DNA replication, thereby locking cells in G1. - ATM/ATR Signaling and DDR Maintenance
Persistent DDR signaling via ATM and ATR is essential for maintaining senescence. These kinases activate downstream effectors such as CHK1/CHK2, and their sustained activation leads to senescence-associated DDR foci, notably γ-H2AX.
C. Epigenetic and Chromatin Changes
Senescent cells undergo significant chromatin remodeling, resulting in transcriptional reprogramming and the suppression of proliferation-associated genes:
- Senescence-Associated Heterochromatin Foci (SAHF)
SAHF are dense chromatin domains enriched in macroH2A, HP1, and H3K9me3. They silence E2F target genes, reinforcing growth arrest. - Histone Modifications
Alterations in histone methylation (e.g., H3K27me3) and acetylation patterns are common, mediated by polycomb group proteins, histone deacetylases, and other chromatin regulators. - Global Transcriptional Reprogramming
Epigenetic alterations lead to upregulation of SASP factors, cell cycle inhibitors, and immune modulators, contributing to the senescence phenotype and its paracrine effects.
III. Senescence-Associated Secretory Phenotype (SASP)
One of the most biologically impactful features of senescent cells is the acquisition of the senescence-associated secretory phenotype (SASP). This phenotype transforms senescent cells from being simply growth-arrested to becoming highly active secretory cells that can profoundly influence their microenvironment. The SASP is a complex and dynamic collection of cytokines, chemokines, growth factors, proteases, and extracellular matrix components that exert both autocrine and paracrine effects.
A. Composition and Components of SASP
Although the exact composition of SASP varies depending on the cell type, senescence trigger, and cellular context, several key molecules are consistently observed:
- Pro-inflammatory cytokines: IL-6, IL-1α, IL-1β, TNF-α
- Chemokines: IL-8 (CXCL8), MCP-1 (CCL2), CXCL1, CCL5
- Growth factors: VEGF, HGF, GM-CSF
- Proteases: Matrix metalloproteinases (MMP-1, MMP-3, MMP-9)
- ECM components and modulators: Fibronectin, osteopontin, plasminogen activator inhibitors (PAI-1)
- Other factors: Reactive oxygen species (ROS), extracellular vesicles (exosomes)
B. Regulation of SASP
The expression of SASP is tightly controlled by multiple signaling pathways and transcription factors:
- NF-κB (Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells)
One of the central regulators of SASP. Activated by persistent DNA damage response (DDR) and cytosolic chromatin fragments, NF-κB drives the transcription of numerous SASP genes. - C/EBPβ (CCAAT/Enhancer Binding Protein Beta)
Cooperates with NF-κB to promote pro-inflammatory SASP gene expression. - mTOR Pathway
mTORC1 activity enhances translation of IL-1α, a key upstream SASP amplifier. Inhibiting mTOR can suppress SASP without reversing senescence. - GATA4 Pathway
GATA4 stabilization due to autophagy inhibition can also activate NF-κB and promote SASP expression.
C. Biological Functions and Impacts of SASP
1. Autocrine Effects
- Reinforces the senescence growth arrest.
- Sustains DDR signaling, particularly via IL-1α and IL-6 feedback loops.
2. Paracrine Effects
- Induces paracrine senescence in neighboring cells (senescence spreading).
- Modulates the tumor microenvironment through:
- Immune surveillance: recruitment of NK cells, T cells, and macrophages to eliminate senescent cells.
- Tissue remodeling and wound healing: via MMPs and growth factors.
3. Tumorigenic Potential
- In a chronic or unresolved context, the SASP can have pro-tumorigenic effects:
- Promotes epithelial-to-mesenchymal transition (EMT).
- Enhances angiogenesis (via VEGF).
- Facilitates immune evasion or creates an immunosuppressive microenvironment.
- Stimulates proliferation of nearby pre-malignant cells.
D. Dynamic and Context-Dependent Nature of SASP
The SASP is not static; its composition and intensity change over time. Early SASP is often dominated by inflammatory cytokines, while late SASP may involve more matrix-remodeling enzymes and growth factors. The cell type and senescence trigger also shape the SASP profile, making it a highly plastic and heterogeneous feature.
Next, we will delve into the methods used to detect and characterize senescent cells, which is crucial for both basic research and clinical translation.
IV. Detection and Biomarkers of Senescent Cells
Identifying senescent cells accurately is essential for both fundamental research and therapeutic targeting in cancer. However, the heterogeneous nature of senescence, and the absence of a single universal marker, make detection a challenging task. Senescence is typically confirmed using a combinatorial approach that includes morphological, molecular, and biochemical markers.
A. Classical Biomarkers
- Senescence-Associated β-Galactosidase (SA-β-Gal) Activity
- The most widely used marker for detecting senescence in vitro and ex vivo.
- Detected at pH 6.0, distinguishing it from lysosomal β-gal activity at acidic pH.
- Reflects lysosomal expansion in senescent cells.
- Limitations: may be absent in certain types of senescence (e.g., non-dividing neurons) or under specific conditions.
- Cell Cycle Inhibitors: p16INK4a and p21CIP1/WAF1
- Elevated expression of these cyclin-dependent kinase inhibitors is a hallmark of senescence.
- p16INK4a marks more irreversible senescence, while p21CIP1 is often associated with early or transient senescence.
- Immunohistochemistry and RT-qPCR are commonly used for detection.
- DNA Damage Markers: γ-H2AX Foci
- Persistent DNA damage foci, especially at telomeres (TIFs: telomere dysfunction-induced foci), indicate chronic DNA damage response (DDR).
- γ-H2AX (phosphorylated histone H2A.X at Ser139) marks double-strand breaks.
- Often co-localizes with 53BP1 in senescent cells.
B. Morphological and Structural Features
- Cellular Hypertrophy and Flattening
- Senescent cells are typically enlarged, flattened, and vacuolated.
- Increased granularity can be observed under phase-contrast microscopy.
- Nuclear Changes
- Altered nuclear morphology, including loss of lamin B1, and formation of senescence-associated heterochromatin foci (SAHF).
- SAHF can be visualized via DAPI staining or specific histone modifications (e.g., H3K9me3).
C. Senescence-Associated Secretory Phenotype (SASP) Markers
- The presence of SASP factors such as IL-6, IL-8, MMP-3, and PAI-1 in the cell culture supernatant or tissue microenvironment can serve as indirect indicators of senescence.
- ELISA, multiplex bead-based assays, and RNA-seq are frequently used to quantify SASP profiles.
D. Emerging and High-Throughput Approaches
- Single-Cell RNA Sequencing (scRNA-seq)
- Reveals transcriptional heterogeneity among senescent cells in tissues.
- Can distinguish senescent subtypes and track temporal SASP evolution.
- Proteomic and Secretomic Profiling
- Mass spectrometry-based analysis can define senescence-specific protein signatures, especially in SASP characterization.
- Fluorescence-Activated Cell Sorting (FACS)
- Uses combinations of cell surface markers, DNA content, and fluorescent probes (e.g., C12FDG for β-Gal activity) to isolate senescent cells.
Next, we will explore how senescence influences cancer development, acting as both a tumor suppressor mechanism and a driver of tumor progression.
V. Cellular Senescence in Cancer — A Double-Edged Sword

Cellular senescence plays a paradoxical role in tumor biology. Initially discovered as a tumor-suppressive mechanism, senescence halts the proliferation of damaged or pre-malignant cells. However, when senescent cells persist and accumulate—especially in the tumor microenvironment—their secretory profile (SASP) can shift toward pro-tumorigenic behavior, promoting immune evasion, metastasis, and therapeutic resistance.
A. Tumor-Suppressive Role of Senescence
1. Barrier to Malignant Transformation
Senescence is activated in response to oncogenic stress, DNA damage, or telomere dysfunction, effectively acting as a fail-safe program to prevent uncontrolled cell proliferation.
- Oncogene-Induced Senescence (OIS): When oncogenes like RAS, BRAF, or MYC are aberrantly activated, senescence is triggered to prevent transformation. This process relies heavily on the p53/p21 and p16/Rb pathways.
- Telomere Shortening and Replicative Senescence: Telomere attrition in the absence of telomerase serves as a checkpoint that halts proliferation of potentially malignant cells.
2. Senescence in Premalignant Lesions
Senescent cells have been identified in early-stage lesions across several cancer types, including:
- Melanocytic nevi, which often harbor activated BRAF mutations yet remain benign due to OIS.
- Prostatic intraepithelial neoplasia (PIN) and colorectal adenomas, where senescence markers are detected in early lesions.
3. Promotion of Immune Surveillance
Senescent cells can recruit and activate immune cells—NK cells, macrophages, CD4+ T cells—via the SASP, facilitating their clearance in a process called senescence surveillance. This contributes to immune-mediated tumor suppression.
B. Tumor-Promoting Role of Senescence
Despite its initial protective function, senescence can become pro-tumorigenic under certain conditions, particularly when senescent cells are not efficiently cleared by the immune system.
1. Chronic SASP and Inflammation
- Long-term SASP secretion promotes chronic inflammation, a well-known cancer hallmark.
- SASP components such as IL-6, IL-8, VEGF, and MMPs support tumorigenesis by:
- Enhancing angiogenesis
- Facilitating epithelial-to-mesenchymal transition (EMT)
- Stimulating cancer cell proliferation and invasion
2. Paracrine Effects on the Tumor Microenvironment
- SASP factors can induce paracrine senescence in stromal cells, altering the tumor stroma to support malignant cell survival.
- Senescent fibroblasts in the tumor microenvironment have been shown to stimulate tumor growth and metastasis, particularly in breast and prostate cancers.
3. Immune Evasion and Tumor Escape
- Chronic SASP may promote recruitment of myeloid-derived suppressor cells (MDSCs) or regulatory T cells (Tregs), fostering an immunosuppressive microenvironment.
- In some cases, SASP factors upregulate PD-L1 expression in cancer cells, contributing to immune evasion.
4. Senescence Bypass and Reprogramming
- Cancer cells may acquire mutations in p53 or Rb pathways, allowing them to escape senescence.
- This escape is often accompanied by genomic instability and acquisition of stem-like properties, increasing malignancy.
- In some models, bypassing senescence has been linked to enhanced metastatic potential and drug resistance.
C. Context Determines Outcome
The impact of senescence in cancer is highly context-dependent:
- Acute, transient senescence may contribute to tumor suppression and tissue repair.
- Chronic, unresolved senescence—especially in aged or immunocompromised tissues—can promote tumorigenesis.
This duality presents a major challenge in the therapeutic exploitation of senescence. Should we promote senescence to halt tumor progression? Or eliminate senescent cells to prevent their deleterious effects? These questions form the basis for the emerging field of senescence-targeted cancer therapy.
In the next section, we will examine therapy-induced senescence (TIS)—a common yet double-edged outcome of conventional cancer treatments like chemotherapy and radiation—and its consequences for long-term cancer control and recurrence.
VI. Therapy-Induced Senescence (TIS)
Conventional cancer treatments such as chemotherapy, radiation therapy, and certain targeted therapies are designed to eliminate rapidly dividing tumor cells by inducing DNA damage and stress. However, rather than triggering apoptosis in all cancer cells, many of these therapies induce a stable growth arrest known as therapy-induced senescence (TIS). While initially considered a favorable outcome, TIS is now understood to be a double-edged sword with significant long-term implications for cancer progression, relapse, and resistance.
A. Mechanisms Underlying TIS
TIS shares core features with other forms of senescence, including permanent cell cycle arrest, DNA damage response activation, and SASP production. Its induction is mediated through several key pathways:
- DNA Damage Response (DDR):
Genotoxic agents such as doxorubicin, etoposide, or ionizing radiation cause double-strand breaks, activating ATM/ATR kinases, leading to p53 stabilization and subsequent p21CIP1 expression. - Oncogene Reactivation and Replication Stress:
Some therapies inadvertently activate oncogenic signaling, leading to oncogene-induced senescence in residual tumor cells. - Epigenetic Remodeling and Chromatin Alterations:
TIS is often accompanied by loss of lamin B1, formation of SAHF, and changes in histone methylation, supporting the senescent phenotype. - Mitochondrial Dysfunction and ROS Accumulation:
Treatment-induced oxidative stress contributes to mitochondrial dysfunction-associated senescence (MiDAS).
B. Functional Consequences of TIS in Cancer
1. Tumor Suppression and Disease Stabilization
- TIS can lead to temporary tumor stasis, allowing the immune system to clear damaged cells.
- In some cases, it contributes to residual disease control and reduces tumor bulk without complete cell elimination.
2. Persistence and Escape from Senescence
- Senescent tumor cells are not dead. Over time, some may escape senescence due to secondary mutations or environmental cues.
- Escapees often exhibit increased genomic instability, stemness, and aggressiveness.
3. SASP-Mediated Tumor Promotion
- TIS-induced SASP includes pro-inflammatory cytokines, growth factors, and proteases, which:
- Promote angiogenesis (e.g., via VEGF)
- Stimulate proliferation of neighboring cancer cells
- Alter stromal and immune cell behavior to favor recurrence
4. Therapy Resistance and Tumor Recurrence
- Senescent cells contribute to the tumor reservoir that survives treatment.
- SASP-mediated remodeling of the tumor microenvironment may drive drug resistance and dormant cell reactivation.
C. Clinical Relevance and Challenges
- Residual senescent cells have been detected in post-treatment biopsies from patients with breast, lung, and prostate cancer.
- Some TIS markers, such as p16INK4a and SA-β-Gal activity, are enriched in treatment-exposed tissues.
- Eliminating these cells might improve long-term outcomes, but selective targeting remains difficult, as some senescent cells also play beneficial roles (e.g., in tissue repair or immune recruitment).
D. Strategies to Modulate TIS Outcomes
- Combining Therapies with Senolytics:
- Administering senolytic drugs post-chemotherapy (e.g., navitoclax, dasatinib + quercetin) may help eliminate therapy-induced senescent cells, reducing recurrence risk.
- Senomorphics to Suppress SASP:
- Using JAK inhibitors, mTOR inhibitors, or NF-κB modulators to blunt the SASP while preserving senescence-induced growth arrest.
- Immunotherapy Synergy:
- Enhancing immune-mediated clearance of TIS cells using checkpoint inhibitors or CAR-T cells targeting senescence-associated antigens.
In the next section, we will delve deeper into therapeutic strategies that directly target senescent cells, including the emerging fields of senolytics and senomorphics
VII. Therapeutic Targeting of Senescence in Cancer
Given the dualistic nature of cellular senescence—protective in early tumor suppression but potentially harmful when senescent cells persist—researchers are developing therapeutic strategies to selectively target senescent cells. Two broad classes of such interventions have emerged: senolytics, which eliminate senescent cells, and senomorphics, which suppress their deleterious secretions without inducing cell death. These approaches represent promising avenues for improving cancer outcomes and reducing therapy-associated complications.
A. Senolytics: Eliminating Senescent Cells
Senolytics are agents designed to selectively induce apoptosis in senescent cells by targeting their unique vulnerabilities. Senescent cells upregulate anti-apoptotic pathways to resist death, making these pathways attractive targets.
1. Mechanism of Action
- Senescent cells often depend on pro-survival proteins such as BCL-2, BCL-xL, and MCL-1.
- Senolytics act by inhibiting these anti-apoptotic factors, thereby tipping the balance toward apoptosis.
2. Key Senolytic Compounds
- Navitoclax (ABT-263): A BCL-2/BCL-xL inhibitor that has shown efficacy in eliminating senescent cells in models of lung and breast cancer, as well as post-chemotherapy.
- Dasatinib + Quercetin: A combinatorial regimen effective in clearing various senescent cell types, including fibroblasts and endothelial cells.
- FOXO4-DRI peptide: Disrupts the interaction between p53 and FOXO4, leading to p53-mediated apoptosis in senescent cells.
- Fisetin: A natural flavonoid with senolytic activity, particularly in preclinical aging and cancer models.
3. Limitations
- Off-target effects: Some senolytics, like navitoclax, cause dose-limiting thrombocytopenia.
- Cell type specificity: Not all senescent cells respond equally to the same agent.
- Incomplete clearance: Residual senescent cells may still exert harmful effects.
B. Senomorphics: Modulating the SASP Without Killing the Cell
Senomorphics aim to neutralize the pro-tumorigenic effects of senescent cells—primarily the SASP—while preserving the beneficial cell cycle arrest.
1. Mechanism of Action
- Senomorphics inhibit SASP transcriptional regulators such as NF-κB, mTOR, and JAK/STAT, thereby reducing the inflammatory and pro-growth signals in the tumor microenvironment.
2. Key Senomorphic Agents
- Rapamycin: Inhibits mTORC1, leading to suppression of SASP translation without reversing cell cycle arrest.
- Ruxolitinib: A JAK1/2 inhibitor that reduces pro-inflammatory cytokines (e.g., IL-6, IL-8) in the SASP.
- Metformin: Activates AMPK, inhibits mTOR, and has shown senomorphic effects in both aging and cancer contexts.
- Glucocorticoids: Such as dexamethasone, known to suppress SASP expression through NF-κB inhibition.
3. Clinical Potential
- Senomorphics may be used adjuvantly after chemotherapy to prevent SASP-induced tumor recurrence or inflammation.
- They offer a safer alternative to senolytics, especially in tissues where complete clearance is not desirable (e.g., liver, skin, vascular endothelium).
C. Emerging Strategies and Combinatorial Approaches
- Immune-Mediated Clearance
- Enhancing immune recognition of senescent cells using immune checkpoint inhibitors, vaccines, or CAR-T cells targeting senescence-associated surface proteins (e.g., DPP4, uPAR).
- Senescence-Inducing + Senolytic Therapies
- Two-step model: Use conventional chemotherapy to induce senescence in tumor cells, followed by senolytics to eliminate them—minimizing long-term SASP-driven effects.
- Biomarker-Guided Therapies
- Identification of senescence-specific surface markers or secretome profiles may enable targeted delivery of senolytics or senomorphics.
D. Challenges and Future Perspectives
- Context specificity: The same senescent cells can be beneficial or harmful depending on the tissue and tumor type.
- Senescence heterogeneity: Not all senescent cells express the same markers, making universal targeting difficult.
- Therapeutic window: Timing of administration (e.g., immediately post-therapy vs. during remission) may determine efficacy and safety.
Next, we’ll explore the experimental models and modern tools used to study senescence, which are essential for developing and validating these therapeutic approaches.
VIII. Experimental Models and Tools to Study Senescence
Understanding the role of cellular senescence in cancer requires reliable experimental models and analytical tools to mimic senescence in vitro and in vivo, dissect its molecular underpinnings, and evaluate therapeutic interventions. Over the past two decades, researchers have developed a diverse array of models and technologies that have significantly advanced senescence biology.
A. In Vitro Models of Senescence
1. Primary Human Fibroblasts
- WI-38, IMR-90, and BJ fibroblasts are widely used to model replicative senescence due to telomere shortening.
- Treatment with hydrogen peroxide, etoposide, or oncogene transduction (e.g., HRASG12V) is used to induce stress- or oncogene-induced senescence.
- These models allow precise control over timing and type of senescence induction.
2. Cancer Cell Lines
- Senescence can be induced in tumor-derived cell lines (e.g., MCF-7, A549, HCT116) using DNA-damaging agents to study therapy-induced senescence (TIS).
- These models are particularly relevant for understanding how senescence contributes to drug resistance, tumor dormancy, or SASP-mediated effects.
3. 3D Cultures and Organoids
- 3D models better recapitulate tissue architecture and microenvironmental factors influencing senescence.
- Organoids derived from primary or tumor tissue can be used to study cell–cell interactions, immune responses, and paracrine senescence.
B. In Vivo Models of Senescence
1. Genetically Engineered Mouse Models (GEMMs)
- Transgenic mice expressing oncogenes (e.g., KRASG12D) or harboring conditional knockouts of p53, p16, or Rb allow study of senescence during tumorigenesis.
- Some models use senescence-reporters, such as p16INK4a-luciferase or SA-β-Gal-tdTomato, to visualize senescent cell burden in vivo.
2. p16-3MR Mouse Model
- A powerful model that expresses a trimodal reporter (3MR) under the control of the p16INK4a promoter.
- Enables non-invasive imaging, drug-induced ablation, or cell sorting of senescent cells using luciferase, thymidine kinase (for GCV-induced death), and red fluorescent protein.
3. Xenograft and Syngeneic Tumor Models
- Tumor cells induced into senescence are transplanted into mice to study SASP-driven tumor promotion, immune clearance, or senolytic responses.
C. Tools for Senescence Detection and Characterization
1. Senescence-Associated β-Galactosidase (SA-β-Gal) Staining
- Still the gold standard for histochemical identification of senescent cells in both cultured cells and tissue sections.
2. Immunohistochemistry and Immunofluorescence
- Used to detect senescence-associated proteins: p16INK4a, p21CIP1, γ-H2AX, 53BP1, and loss of Lamin B1.
- Co-staining with proliferation markers (e.g., Ki-67) helps distinguish senescent from quiescent or differentiated cells.
3. Flow Cytometry-Based Approaches
- Uses fluorescent SA-β-Gal substrates (e.g., C12FDG), combined with DNA content analysis and surface markers to quantify and sort senescent cells.
4. Transcriptomic and Epigenomic Profiling
- Bulk RNA-seq and single-cell RNA-seq (scRNA-seq) are used to map senescence transcriptional states and SASP heterogeneity.
- ATAC-seq and ChIP-seq reveal chromatin remodeling during senescence, including formation of senescence-associated heterochromatin foci (SAHF).
5. Proteomics and Secretomics
- Mass spectrometry identifies SASP components and senescence-enriched proteins.
- Useful for discovering biomarkers and designing senescence-targeted therapies.
D. High-Throughput Screening for Senescence Modulators
- CRISPR-Cas9 screens identify genes involved in senescence induction, maintenance, or escape.
- Drug libraries are screened for senolytic or senomorphic properties using automated imaging and cell viability assays.
E. Limitations and Considerations
- Species-specific differences: Senescence markers and responses in mice may not fully reflect human biology.
- Artificial induction: High doses of DNA damage or forced oncogene expression may not accurately replicate physiological senescence.
- Cell type variability: Not all cells undergo or express senescence markers uniformly—highlighting the importance of using multiple readouts.
Conclusion
Cellular senescence is a complex and multifaceted process that plays both protective and harmful roles in cancer. While it serves as a critical barrier against tumorigenesis, the persistence of senescent cells and their SASP can promote cancer progression, immune evasion, and therapy resistance. Advances in experimental models and senescence-targeted therapies—including senolytics and senomorphics—offer promising strategies to modulate this double-edged phenomenon. As our understanding deepens, integrating senescence into cancer diagnostics and treatment may open new avenues for improving patient outcomes and preventing relapse.
References
- Campisi, J. (2013). Aging, cellular senescence, and cancer. Annual Review of Physiology, 75, 685–705.
https://doi.org/10.1146/annurev-physiol-030212-183653 - He, S., & Sharpless, N. E. (2017). Senescence in Health and Disease. Cell, 169(6), 1000–1011.
https://doi.org/10.1016/j.cell.2017.05.015 - Di Micco, R., Fumagalli, M., & d’Adda di Fagagna, F. (2021). Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nature Reviews Molecular Cell Biology, 22(2), 75–95.
https://doi.org/10.1038/s41580-020-00314-w - Coppe, J. P., Desprez, P. Y., Krtolica, A., & Campisi, J. (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology, 5, 99–118.
https://doi.org/10.1146/annurev-pathol-121808-102144 - Childs, B. G., Gluscevic, M., Baker, D. J., Laberge, R. M., Marquess, D., Dananberg, J., & van Deursen, J. M. (2017). Senescent cells: an emerging target for diseases of ageing. Nature Reviews Drug Discovery, 16(10), 718–735.
https://doi.org/10.1038/nrd.2017.116 - Hernandez-Segura, A., Nehme, J., & Demaria, M. (2018). Hallmarks of Cellular Senescence. Trends in Cell Biology, 28(6), 436–453.
https://doi.org/10.1016/j.tcb.2018.02.001 - Gorgoulis, V., Adams, P. D., Alimonti, A., Bennett, D. C., Bischof, O., Bishop, C., Campisi, J., Collado, M., Evangelou, K., Ferbeyre, G., et al. (2019). Cellular Senescence: Defining a Path Forward. Cell, 179(4), 813–827.
https://doi.org/10.1016/j.cell.2019.10.005 - Wang, C., Jurk, D., Maddick, M., Nelson, G., Martin-Ruiz, C., & von Zglinicki, T. (2009). DNA damage response and cellular senescence in tissues of aging mice. Aging Cell, 8(3), 311–323.
https://doi.org/10.1111/j.1474-9726.2009.00481.x - Wiley, C. D., & Campisi, J. (2021). From ancient pathways to aging cells – connecting metabolism and cellular senescence. Cell Metabolism, 33(9), 1676–1696.
DOI: 10.1016/j.cmet.2016.05.010 - Naylor, R. M., Baker, D. J., & van Deursen, J. M. (2013). Senescent cells: a novel therapeutic target for aging and age-related diseases. Clinical Pharmacology & Therapeutics, 93(1), 105–116.
https://doi.org/10.1038/clpt.2012.193 - Tchkonia, T., Zhu, Y., van Deursen, J., Campisi, J., & Kirkland, J. L. (2013). Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. The Journal of Clinical Investigation, 123(3), 966–972.
https://doi.org/10.1172/JCI64098 - Demaria, M., O’Leary, M. N., Chang, J., Shao, L., Liu, S., Alimirah, F., Koenig, K., Le, C., Mitin, N., Deal, A. M., et al. (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discovery, 7(2), 165–176.
https://doi.org/10.1158/2159-8290.CD-16-0241 - Childs, B. G., Baker, D. J., Kirkland, J. L., Campisi, J., & van Deursen, J. M. (2017). Senescence and apoptosis: dueling or complementary cell fates? EMBO Reports, 18(11), 1573–1581.
https://doi.org/10.15252/embr.201439245 - Schmitt, C.A., Wang, B. & Demaria, M. Senescence and cancer — role and therapeutic opportunities. Nat Rev Clin Oncol 19, 619–636 (2022).
https://doi.org/10.1038/s41571-022-00668-4 - Wiley, C. D., Velarde, M. C., Lecot, P., Liu, S., Sarnoski, E. A., Freund, A., Shirakawa, K., Lim, H. W., Davis, S. S., Ramanathan, A., et al. (2016). Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metabolism, 23(2), 303–314.
https://doi.org/10.1016/j.cmet.2015.11.011

{kind=link}